
New activation mechanism for half-sandwich organometallic anticancer complexes†
 ‡a
Joan J.
Soldevila-Barreda,
‡a
Juliusz A.
Wolny,
‡b
Christopher A.
Wootton,
a
Abraha
Habtemariam,
a
Isolda
Romero-Canelón,
a
Feng
Chen,
a
Guy J.
Clarkson,
a
Peter B.
O'Connor,
a
Volker
Schünemann
b
and
Peter J.
Sadler
*a
‡a
Joan J.
Soldevila-Barreda,
‡a
Juliusz A.
Wolny,
‡b
Christopher A.
Wootton,
a
Abraha
Habtemariam,
a
Isolda
Romero-Canelón,
a
Feng
Chen,
a
Guy J.
Clarkson,
a
Peter B.
O'Connor,
a
Volker
Schünemann
b
and
Peter J.
Sadler
*a
Abstract
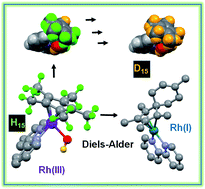
The Cpx C–H protons in certain organometallic RhIII half-sandwich anticancer complexes [(η5-Cpx)Rh(N,N′)Cl]+, where Cpx = Cp*, phenyl or biphenyl-Me4Cp, and N,N′ = bipyridine, dimethylbipyridine, or phenanthroline, can undergo rapid sequential deuteration of all 15 Cp* methyl protons in aqueous media at ambient temperature. DFT calculations suggest a mechanism involving abstraction of a Cp* proton by the Rh–hydroxido complex, followed by sequential H/D exchange, with the Cp* rings behaving like dynamic molecular ‘twisters’. The calculations reveal the crucial role of pπ orbitals of N,N′-chelated ligands in stabilizing deprotonated Cpx ligands, and also the accessibility of RhI–fulvene intermediates. They also provide insight into why biologically-inactive complexes such as [(Cp*)RhIII(en)Cl]+ and [(Cp*)IrIII(bpy)Cl]+ do not have activated Cp* rings. The thiol tripeptide glutathione (γ-L-Glu-L-Cys-Gly, GSH) and the activated dienophile N-methylmaleimide, (NMM) did not undergo addition reactions with the proposed RhI–fulvene, although they were able to control the extent of Cp* deuteration. We readily trapped and characterized RhI–fulvene intermediates by Diels–Alder [4+2] cyclo-addition reactions with the natural biological dienes isoprene and conjugated (9Z,11E)-linoleic acid in aqueous media, including cell culture medium, the first report of a Diels–Alder reaction of a metal-bound fulvene in aqueous solution. These findings will introduce new concepts into the design of organometallic Cp* anticancer complexes with novel mechanisms of action.
 Please wait while we load your content...
Please wait while we load your content...

