
Quantum chemical studies of redox properties and conformational changes of a four-center iron CO2 reduction electrocatalyst†

Abstract
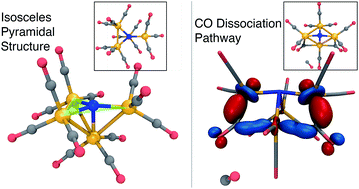
The CO2 reduction electrocatalyst [Fe4N(CO)12]− (abbrev. 1−) reduces CO2 to HCO2− in a two-electron, one-proton catalytic cycle. Here, we employ ab initio calculations to estimate the first two redox potentials of 1− and explore the pathway of a side reaction involving CO dissociation from 13−. Using the BP86 density functional approximation, the redox potentials were computed with a root mean squared error of 0.15 V with respect to experimental data. High temperature Born–Oppenheimer molecular dynamics was employed to discover a reaction pathway of CO dissociation from 13− with a reaction energy of +10.6 kcal mol−1 and an activation energy of 18.8 kcal mol−1; including harmonic free energy terms, this yields ΔGsep = 1.4 kcal mol−1 for fully separated species and ΔG‡ = +17.4 kcal mol−1, indicating CO dissociation is energetically accessible at ambient conditions. The analogous dissociation pathway from 12− has a reaction energy of 22.1 kcal mol−1 and an activation energy of 22.4 kcal mol−1 (ΔGsep = 12.8 kcal mol−1, ΔG‡ = +18.1 kcal mol−1). Our computed harmonic vibrational analysis of [Fe4N(CO)11]3− or 23− reveals a distinct CO-stretching peak red-shifted from the main CO-stretching band, pointing to a possible vibrational signature of dissociation. Multi-reference CASSCF calculations are used to check the assumptions of the density functional approximations that were used to obtain the majority of the results.
 Please wait while we load your content...
Please wait while we load your content...

