
Fast and accurate prediction of the regioselectivity of electrophilic aromatic substitution reactions†
 a
Jan H.
Jensen,
*a
a
Jan H.
Jensen,
*a
Abstract
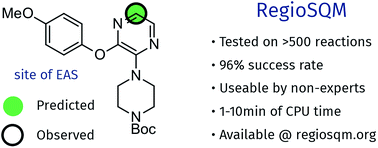
While computational prediction of chemical reactivity is possible it usually requires expert knowledge and there are relatively few computational tools that can be used by a bench chemist to help guide synthesis. The RegioSQM method for predicting the regioselectivity of electrophilic aromatic substitution reactions of heteroaromatic systems is presented in this paper. RegioSQM protonates all aromatic C–H carbon atoms and identifies those with the lowest free energies in chloroform using the PM3 semiempirical method as the most nucleophilic center. These positions are found to correlate qualitatively with the regiochemical outcome in a retrospective analysis of 96% of more than 525 literature examples of electrophilic aromatic halogenation reactions. The method is automated and requires only a SMILES string of the molecule of interest, which can easily be generated using chemical drawing programs such as ChemDraw. The computational cost is 1–10 minutes per molecule depending on size, using relatively modest computational resources and the method is freely available via a web server at http://www.regiosqm.org. RegioSQM should therefore be of practical use in the planning of organic synthesis.
- This article is part of the themed collection: The ChemRxiv Collection
 Please wait while we load your content...
Please wait while we load your content...

