
Computational modelling of solvent effects in a prolific solvatomorphic porous organic cage†

Abstract
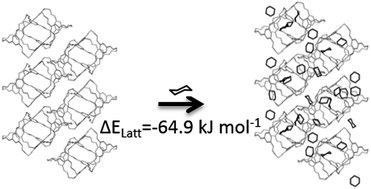
Crystal structure prediction methods can enable the in silico design of functional molecular crystals, but solvent effects can have a major influence on relative lattice energies, sometimes thwarting predictions. This is particularly true for porous solids, where solvent included in the pores can have an important energetic contribution. We present a Monte Carlo solvent insertion procedure for predicting the solvent filling of porous structures from crystal structure prediction landscapes, tested using a highly solvatomorphic porous organic cage molecule, CC1. Using this method, we can understand why the predicted global energy minimum structure for CC1 is never observed from solvent crystallisation. We also explain the formation of three different solvatomorphs of CC1 from three structurally-similar chlorinated solvents. Calculated solvent stabilisation energies are found to correlate with experimental results from thermogravimetric analysis, suggesting a future computational framework for a priori materials design that factors in solvation effects.
- This article is part of the themed collection: Methods and applications of crystal structure prediction
 Please wait while we load your content...
Please wait while we load your content...

