
Cooperative bimetallic reactivity of a heterodinuclear molybdenum–copper model of Mo–Cu CODH†
 b
and
Stanislav
Groysman
*a
b
and
Stanislav
Groysman
*a
Abstract
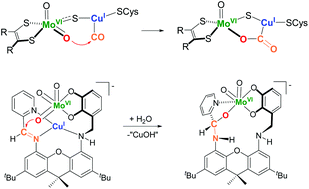
The synthesis of a heterodinucleating ligand LH2 (LH2 = (E)-3-(((2,7-di-tert-butyl-9,9-dimethyl-5-((pyridin-2-ylmethylene)amino)-9H-xanthen-4-yl)amino)methyl)benzene-1,2-diol) was undertaken toward a functional model of the bimetallic active site found in Mo–Cu carbon monoxide dehydrogenase (Mo–Cu CODH), and to understand the origins of heterobimetallic cooperativity exhibited by the enzyme. LH2 features a hard potentially dianionic catechol chelate for binding Mo(VI) and a soft iminopyridine chelate for binding Cu(I). Treatment of LH2 with either Cu(I) or M(VI) (M = Mo, W) sources leads to the anticipated site-selective incorporation of the respective metals. While both [CuI(LH2)]+ and [MVIO3(L)]2− complexes are stable in the solid state, [MVIO3(L)]2− complexes disproportionate in solution to give [MVIO2(L)2](NEt4)2 complexes, with [MVIO4]2− as the by-product. The incorporation of BOTH Mo(VI) and Cu(I) into L forms a highly reactive heterobimetallic complex [MoVIO3CuI(L)](NEt4)2, whose formation and reactivity was interrogated via1H NMR/UV-vis spectroscopy and DFT calculations. These studies reveal that the combination of the two metals triggers oxidation reactivity, in which a nucleophilic Mo(VI) trioxo attacks Cu(I)-bound imine. The major product of the reaction is a crystallographically characterized molybdenum(VI) complex [Mo(L′)O2](NEt4) coordinated by a modified ligand L′ that contains a new C–O bond in place of the imine functionality. This observed hydroxylation reactivity is consistent with the postulated first step of Mo–Cu CODH (nucleophilic attack of the Mo(VI)–oxo on the Cu(I)-bound electrophilic CO) and xanthine oxidoreductase (nucleophilic attack of Mo(VI)–oxo on the electrophilic xanthine carbon).
 Please wait while we load your content...
Please wait while we load your content...

