
Automated calculation of thermal rate coefficients using ring polymer molecular dynamics and machine-learning interatomic potentials with active learning
 *bc
and
*bc
and
Abstract
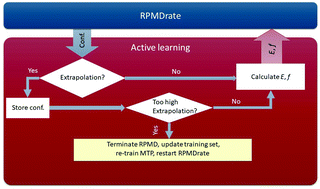
We propose a methodology for the fully automated calculation of thermal rate coefficients of gas phase chemical reactions, which is based on combining ring polymer molecular dynamics (RPMD) and machine-learning interatomic potentials actively learning on-the-fly. Based on the original computational procedure implemented in the RPMDrate code, our methodology gradually and automatically constructs the potential energy surfaces (PESs) from scratch with the data set points being selected and accumulated during the RPMDrate simulation. Such an approach ensures that our final machine-learning model provides a reliable description of the PES that avoids artifacts during exploration of the phase space by RPMD trajectories. We tested our methodology on two representative thermally activated chemical reactions studied recently by RPMDrate at temperatures within the interval of 300–1000 K. The corresponding PESs were generated by fitting to only a few thousand automatically generated structures (less than 5000) while the RPMD rate coefficients showed deviation from the reference values within the typical convergence error of RPMDrate. In future, we plan to apply our methodology to chemical reactions that proceed via complex-formation thus providing a completely general tool for calculating RPMD thermal rate coefficients for any polyatomic gas phase chemical reaction.
 Please wait while we load your content...
Please wait while we load your content...

