
Pore-filling contamination in metal–organic frameworks†
 a
and
Elena
Besley
*a
a
and
Elena
Besley
*a
Abstract
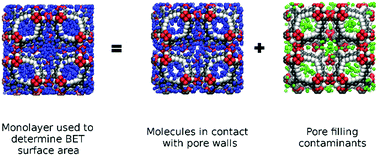
Tuneable pore sizes, ordered crystal structures, and large surface areas are some of the main attractive features of metal–organic frameworks (MOFs). To fully understand the structure–property relationships of these materials, accurate characterisation of their structural features is essential. The surface areas of MOFs are routinely estimated from the physical adsorption of gases. By applying the Brunauer, Emmett & Teller (BET) theory to an adsorption isotherm, the surface area is calculated from the amount of gas that forms a monolayer on the pore surface. While this technique is used ubiquitously within the porous solid community, its accuracy can be greatly affected by pore-filling contamination. This process causes an overestimation of the BET surface area from the overlap of surface and pore-filling adsorption as molecules that are not in contact with the surface are erroneously included into the surface area calculation. Experimentally, it is rather challenging to examine the effects of pore-filling contamination, which typically rely on accurate atomistic simulations to provide insight. In this work, we employ grand canonical Monte Carlo simulations and other theoretical approaches to assess the impact of pore-filling contamination on MOF surface areas. With a focus on the rht and nbo topologies, we show how experimental studies that suggest MOF surface areas can be increased by replacing phenyl rings for alkynes are largely influenced by the pore-filling contamination effect.
 Please wait while we load your content...
Please wait while we load your content...

