
The dioxygen adducts of iron and manganese porphyrins: electronic structure and binding energy†
 a
and
Kristine
Pierloot
*a
a
and
Kristine
Pierloot
*a
Abstract
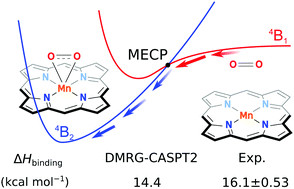
In this paper, we present a thorough study of the electronic structures and binding energies of O2 to iron and manganese porphyrins (FeP and MnP), employing a state-of-the-art computational technique known as second-order perturbation theory based on density matrix renormalization group (DMRG-CASPT2). By investigating an extensive list of different binding modes and spin states, we provide a clear and conclusive description of the ground state of MnP–O2, confirming available experimental evidences. Our results show that MnP–O2 favours a side-on quartet structure, with strong charge transfer between MnP and O2. We also calculated the standard binding enthalpies of O2 to different metal porphyrins and showed that an agreement between calculated results and experimental data to within 2 kcal mol−1 can be achieved. Our calculations confirm the experimental observation that the binding of O2 to manganese porphyrin is stronger by around 4–6 kcal mol−1 than to the corresponding ferrous porphyrin.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

