
Solvent effects on the decarboxylation of trichloroacetic acid: insights from ab initio molecular dynamics simulations†

Abstract
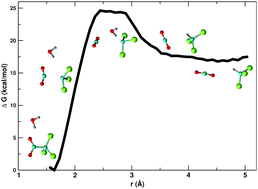
The kinetics of trichloroacetic acid (TCA) decarboxylation strongly depends on the solvent in which it occurs, proceeding faster in polar aprotic solvents compared to protic solvents. In particular, the reaction is known to be fast in DMSO even at room temperature and is rather slow in water even at higher temperatures. In order to understand the role of the solvent in the kinetics of TCA decarboxylation, the present study investigates this reaction using both ab initio molecular dynamics (AIMD) simulations in explicit solvents and static electronic structure calculations with the SMD polarizable continuum model, considering DMSO and water as solvents. Both methodologies yield activation free energies in good agreement with experimental data, however they differ with respect to the reaction profile for the process occurring in water. The simulations suggest that DMSO does not participate chemically in the reaction and that the high reaction rate in DMSO can be explained by differential solvation of the reactant and transition state. In water, a protonation step was observed along the simulation trajectory, indicating chemical participation of the solvent in this case. Moreover, the continuum model has shown to be useful to predict the reaction rates in other solvents, suggesting that reaction rates increase upon decreasing solvent polarity up to the point where the apolar solvents are not able to efficiently screen the strong electrostatic interactions to form the required isolated ionic species.
 Please wait while we load your content...
Please wait while we load your content...

