
Quantitative description of 1H SQ and DQ coherences for the hydroxyl disorder within hydrous ringwoodite†

Abstract
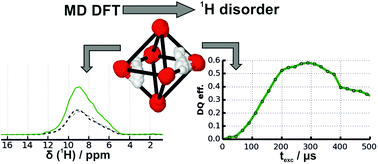
Proton-containing point defects in solid materials are important for a variety of properties ranging from ionic transport over thermal conductivity up to compressibility. Ultrafast magic-angle spinning techniques nowadays offer high-resolution solid-state NMR spectra, even for 1H, and thus open up possibilities to study the underlying defect chemistry. Nevertheless, disorder within such defects again leads to heavy spectral overlap of 1H resonances, which prevents quantitative analysis of defect concentrations, if several defect types are present. Here, we present a strategy to overcome this limitation by simulating the 1H lineshape as well as 1H–1H double-quantum buildup curves, which we then validate against the experimental data in a joint cost function. To mimic the local structural disorder, we use molecular dynamics simulations at the DFT level. It turned out to be advantageous for the joint refinement to put the computational effort into the structural optimisation to derive accurate proton positions and to use empirical correlations for the relation between isotropic and anisotropic 1H chemical shifts and structural elements. The expressiveness of this approach is demonstrated on ringwoodite's (γ-Mg2SiO4) OH defect chemistry containing four different defect types in octahedral and tetrahedral voids with both pure Mg and mixed Si and Mg cation environments. Still, we determine the ratio for each defect type with an accuracy of about 5% as a result of the minimization of the joint cost function. We expect that our approach is generally applicable for local proton disorder and might prove to be a valuable alternative to the established AIRSS and Monte Carlo methods, respectively.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

