
Accurate theoretical characterization of dioxygen difluoride: a problem resolved
 *ab
Alexander
Alijah,
*b
Thibaud
Cours,
b
Nejm-Eddine
Jaidane
a
and
Najoua
Derbel
*a
*ab
Alexander
Alijah,
*b
Thibaud
Cours,
b
Nejm-Eddine
Jaidane
a
and
Najoua
Derbel
*a
Abstract
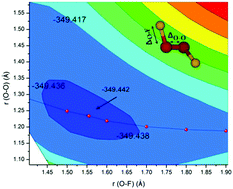
Dioxygen difluoride is a tough molecule that has defied accurate theoretical description for many decades. In the present work we have identified the reason for this resistance: the flatness of the OO, and more important OF, stretching potential energy curves, which make it difficult to localise the global minimum. It is not related to the weak multi-reference character. Using high-level CCSD(T)-F12/VTZ-F12 ab initio theory, the global minimum has been properly located and vibrationally averaged bond lengths obtained. These vibrationally averaged parameters agree with experimental data to within 0.01 Å. Averaging was found essential to achieve this unprecedented accuracy. We have then simulated the IR and UV spectra, which compare well with experimental data and permit identification of the observed transitions.
 Please wait while we load your content...
Please wait while we load your content...

