
A combined theoretical and experimental investigation of the kinetics and dynamics of the O(1D) + D2 reaction at low temperature†

Abstract
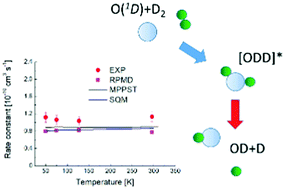
The O(1D) + H2 reaction is a prototype for simple atom–diatom insertion type mechanisms considered to involve deep potential wells. While exact quantum mechanical methods can be applied to describe the dynamics, such calculations are challenging given the numerous bound quantum states involved. Consequently, efforts have been made to develop alternative theoretical strategies to portray accurately the reactive process. Here we report an experimental and theoretical investigation of the O(1D) + D2 reaction over the 50–296 K range. The calculations employ three conceptually different approaches – mean potential phase space theory, the statistical quantum mechanical method and ring polymer molecular dynamics. The calculated rate constants are in excellent agreement over the entire temperature range, exhibiting only weak temperature dependence. The agreement between experiment and theory is also very good, with discrepancies smaller than 26%. Taken together, the present and previous theoretical results validate the hypothesis that long-lived complex formation dominates the reaction dynamics at low temperature.
 Please wait while we load your content...
Please wait while we load your content...

