
Conformational chiral polymorphism in cis-bis-triphenylphosphine complexes of transition metals†

Abstract
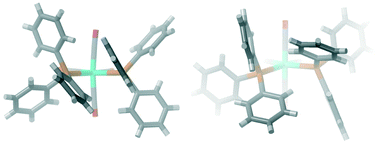
The structure of cis-[Mo(CO)4(PPh3)2] 1 was determined by F. A. Cotton, D. J. Darensbourg, S. Klein and B. W. S. Kolthammer, Inorg. Chem., 1982, 21, 1651–1655, with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A second polymorph 2 is reported here, with the space group P21/c. The compounds differ in the interactions between the conformational chiral triphenylphosphine groups. In 1, there is π–π stacking between adjacent phenyl groups, whereas in 2, there are σ–π interactions instead. A search of the Cambridge Structural Database reveals that this is a relatively frequent occurrence in cis-bis-triphenylphosphine complexes and the phenomenon can be analysed by means of the C(ipso)–P–M–P torsion angles. The majority of compounds fall in the π–π stacking data area with torsion angles of 10–15° and 55–60°; however, for octahedrally coordinated metals, the optimum is a σ–π interaction at 40°/40°. This corresponds well to the values in 2: 46°/40°, but for 1, we instead find the torsion angles to be 11°/18°. There is indeed a small occurrence of these values as well in the data, and it appears that for 1, this conformation is stabilised by weak CO⋯H–C hydrogen bonds. Density functional theory (DFT) calculations indicate that 1 is the more stable polymorph by 72 kJ mol−1 but that the strain of the complexes (the difference between a relaxed molecule in the respective conformation and the structure in the crystal) is larger for 1 than for 2, further indicating that a special intermolecular interaction is responsible for the stability of this polymorph. In both polymorphs, the triphenylphosphines have the same conformational chirality, consistent with single-molecule calculations that predict racemic conformations to be substantially higher in energy for both σ–π interactions (+17 kJ mol−1) and π–π stacking (+30 kJ mol−1).
. A second polymorph 2 is reported here, with the space group P21/c. The compounds differ in the interactions between the conformational chiral triphenylphosphine groups. In 1, there is π–π stacking between adjacent phenyl groups, whereas in 2, there are σ–π interactions instead. A search of the Cambridge Structural Database reveals that this is a relatively frequent occurrence in cis-bis-triphenylphosphine complexes and the phenomenon can be analysed by means of the C(ipso)–P–M–P torsion angles. The majority of compounds fall in the π–π stacking data area with torsion angles of 10–15° and 55–60°; however, for octahedrally coordinated metals, the optimum is a σ–π interaction at 40°/40°. This corresponds well to the values in 2: 46°/40°, but for 1, we instead find the torsion angles to be 11°/18°. There is indeed a small occurrence of these values as well in the data, and it appears that for 1, this conformation is stabilised by weak CO⋯H–C hydrogen bonds. Density functional theory (DFT) calculations indicate that 1 is the more stable polymorph by 72 kJ mol−1 but that the strain of the complexes (the difference between a relaxed molecule in the respective conformation and the structure in the crystal) is larger for 1 than for 2, further indicating that a special intermolecular interaction is responsible for the stability of this polymorph. In both polymorphs, the triphenylphosphines have the same conformational chirality, consistent with single-molecule calculations that predict racemic conformations to be substantially higher in energy for both σ–π interactions (+17 kJ mol−1) and π–π stacking (+30 kJ mol−1).
- This article is part of the themed collection: Introducing the CrystEngComm Advisory Board and their research
 Please wait while we load your content...
Please wait while we load your content...

