
Coarse-grained molecular dynamics studies of the structure and stability of peptide-based drug amphiphile filaments†

Abstract
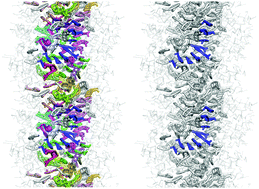
Peptide-based supramolecular filaments, in particular filaments self-assembled by drug amphiphiles (DAs), possess great potential in the field of drug delivery. These filaments possess one hundred percent drug loading, with a release mechanism that can be tuned based on the dissociation of the supramolecular filaments and the degradation of the DAs [Cheetham et al., J. Am. Chem. Soc., 2013, 135(8), 2907]. Recently, much attention has been drawn to the competing intermolecular interactions that drive the self-assembly of peptide-based amphiphiles into supramolecular filaments. Recently, we reported on long-time atomistic molecular dynamics simulations to characterize the structure and growth of chiral filaments by the self-assembly of a DA containing the aromatic anti-cancer drug camptothecin [Kang et al., Macromolecules, 2016, 49(3), 994]. We found that the π–π stacking of the aromatic drug governs the early stages of the self-assembly process, while also contributing towards the chirality of the self-assembled filament. Based on these all-atomistic simulations, we now build a chemically accurate coarse-grained model that can capture the structure and stability of these supramolecular filaments at long time-scales (microseconds). These coarse-grained models successfully recapitulate the growth of the molecular clusters (and their elongation trends) compared with previously reported atomistic simulations. Furthermore, the interfacial structure and the helicity of the filaments are conserved. Next, we focus on characterization of the disassembly process of a 0.675 μm DA filament at microsecond time-scales. These results provide very useful tools for the rational design of functional supramolecular filaments, in particular supramolecular filaments for drug delivery applications.
 Please wait while we load your content...
Please wait while we load your content...

