
Nucleophilic addition and substitution at coordinatively saturated boron by facile 1,2-hydrogen shuttling onto a carbene donor†
 ab
Merle
Arrowsmith,
ab
Holger
Braunschweig,
*ab
Rian D.
Dewhurst,
ab
J. Oscar C.
Jiménez-Halla
ac
and
ab
Merle
Arrowsmith,
ab
Holger
Braunschweig,
*ab
Rian D.
Dewhurst,
ab
J. Oscar C.
Jiménez-Halla
ac
and
Abstract
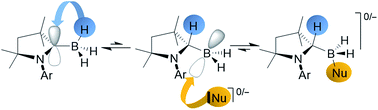
The reaction of [(cAACMe)BH3] (cAACMe = 1-(2,6-iPr2C6H3)-3,3,5,5-tetramethylpyrrolidin-2-ylidene) with a range of organolithium compounds led to the exclusive formation of the corresponding (dihydro)organoborates, Li+[(cAACMeH)BH2R]− (R = sp3-, sp2-, or sp-hybridised organic substituent), by migration of one boron-bound hydrogen atom to the adjacent carbene carbon of the cAAC ligand. A subsequent deprotonation/salt metathesis reaction with Me3SiCl or spontaneous LiH elimination yielded the neutral cAAC-supported mono(organo)boranes, [(cAACMe)BH2R]. Similarly the reaction of [(cAACMe)BH3] with a neutral donor base L resulted in adduct formation by shuttling one boron-bound hydrogen to the cAAC ligand, to generate [(cAACMeH)BH2L], either irreversibly (L = cAACMe) or reversibly (L = pyridine). Variable-temperature NMR data and DFT calculations on [(cAACMeH)BH2(cAACMe)] show that the hydrogen on the former carbene carbon atom exchanges rapidly with the boron-bound hydrides.
 Please wait while we load your content...
Please wait while we load your content...

