
Identifying the first folded alkylbenzene via ultraviolet, infrared, and Raman spectroscopy of pentylbenzene through decylbenzene†
 a
Sebastian
Bocklitz,
b
Daniel P.
Tabor,
c
Edwin L.
Sibert III,
*c
Martin A.
Suhm
b
and
Timothy S.
Zwier
*a
a
Sebastian
Bocklitz,
b
Daniel P.
Tabor,
c
Edwin L.
Sibert III,
*c
Martin A.
Suhm
b
and
Timothy S.
Zwier
*a
Abstract
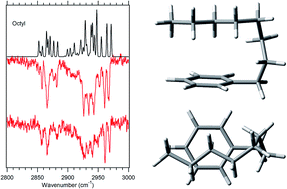
The conformational preferences of pentyl- through decylbenzene are studied under jet-cooled conditions in the gas phase. Laser-induced fluorescence excitation spectra, fluorescence-dip infrared spectra in the alkyl CH stretch region, and Raman spectra are combined to provide assignments for the observed conformers. Density functional theory calculations at the B3LYP-D3BJ/def2TZVP level of theory provide relative energies and normal mode vibrations that serve as inputs for an anharmonic local mode theory introduced in earlier work on alkylbenzenes with n = 2–4. This model explicitly includes anharmonic mixing of the CH stretch modes with the overtones of scissors/bend modes of the CH2 and CH3 groups in the alkyl chain, and is used to assign and interpret the single-conformation IR spectra. In octylbenzene, a pair of LIF transitions shifted −92 and −78 cm−1 from the all-trans electronic origin have unique alkyl CH stretch transitions that are fit by the local model to a g1g3g4 conformation in which the alkyl chain folds back over the aromatic ring π cloud. Its calculated energy is only 1.0 kJ mol−1 above the all-trans global minimum. This fold is at an alkyl chain length less than half that of the pure alkanes (n = 18), consistent with a smaller energy cost for the g1 dihedral and the increased dispersive interaction of the chain with the π cloud. Local site frequencies for the entire set of conformers from the local mode model show ‘edge effects’ that raise the site frequencies of CH2(1) and CH2(2) due to the phenyl ring and CH2(n − 1) due to the methyl group. The g1g3g4 conformer also shows local sites shifted up in frequency at CH2(3) and CH2(6) due to interaction with the π cloud.
 Please wait while we load your content...
Please wait while we load your content...

