
Photoinduced electron transfer from rylenediimide radical anions and dianions to Re(bpy)(CO)3 using red and near-infrared light†
 ‡a
Jose F.
Martinez,
‡a
Svante
Hedström,
b
Brian T.
Phelan,
a
Michael R.
Wasielewski
*a
‡a
Jose F.
Martinez,
‡a
Svante
Hedström,
b
Brian T.
Phelan,
a
Michael R.
Wasielewski
*a
Abstract
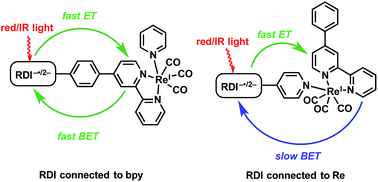
A major goal of artificial photosynthesis research is photosensitizing highly reducing metal centers using as much as possible of the solar spectrum reaching Earth's surface. The radical anions and dianions of rylenediimide (RDI) dyes, which absorb at wavelengths as long as 950 nm, are powerful photoreductants with excited state oxidation potentials that rival or exceed those of organometallic chromophores. These dyes have been previously incorporated into all-organic donor–acceptor systems, but have not yet been shown to reduce organometallic centers. This study describes a set of dyads in which perylenediimide (PDI) or naphthalenediimide (NDI) chromophores are attached to Re(bpy)(CO)3 through either the bipyridine ligand or more directly to the Re center via a pyridine ligand. The chromophores are reduced with a mild reducing agent, after which excitation with long-wavelength red or near-infrared light leads to reduction of the Re complex. The kinetics of electron transfer from the photoexcited anions to the Re complex are monitored using transient visible/near-IR and mid-IR spectroscopy, complemented by theoretical spectroscopic assignments. The photo-driven charge shift from the reduced PDI or NDI to the complex occurs in picoseconds regardless of whether PDI or NDI is attached to the bipyridine or to the Re center, but back electron transfer is found to be three orders of magnitude slower with the chromophore attached to the Re center. These results will inform the design of future catalytic systems that incorporate RDI anions as chromophores.
- This article is part of the themed collection: Celebrating the 2017 RSC Prize and Award Winners
 Please wait while we load your content...
Please wait while we load your content...

