
Understanding the atomic-level process of CO-adsorption-driven surface segregation of Pd in (AuPd)147 bimetallic nanoparticles†
 *a
*a
Abstract
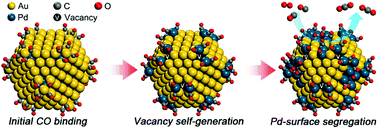
When the elements that compose bimetallic catalysts interact asymmetrically with reaction feedstock, the surface concentration of the bimetallic catalysts and the morphology of the reaction center evolve dynamically as a function of environmental factors such as the partial pressure of the triggering molecule. Relevant experimental and theoretical findings of the dynamic structural evolution of bimetallic catalysts under the reaction conditions are emerging, thus enabling the design of more consistent, reliable, and efficient bimetallic catalysts. In an initial attempt to provide an atomic-level understanding of the adsorption-induced structural evolution of bimetallic nanoparticles (NPs) under CO oxidation conditions, we used density functional theory to study the details of CO-adsorption-driven Pd surface segregation in (AuPd)147 bimetallic NPs. The strong CO affinity of Pd provides a driving force for Pd surface segregation. We found that the vertex site of the NP becomes a gateway for the initial Pd–Au swapping and the subsequent formation of an internal vacancy. This self-generated internal vacancy easily diffuses inside the NP and activates Pd–Au swapping pathways in the (100) NP facet. Our results reveal how the surface and internal concentrations of bimetallic NPs respond immediately to changes in the reaction conditions. Our findings should aid in the rational design of highly active and versatile bimetallic catalysts by considering the environmental factors that systematically affect the structure of bimetallic catalysts under the reaction conditions.
 Please wait while we load your content...
Please wait while we load your content...

