
Synthesis of novel pentacyclic triterpene–Neu5Ac2en derivatives and investigation of their in vitro anti-influenza entry activity†‡
 *a
*a
Abstract
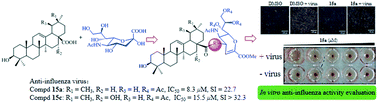
Sialic acid derivatives, analogs, and their conjugates are important pharmacophores. Modification of the C-4 hydroxyl group of sialic acid can lead to derivatives, such as zanamivir, with potent anti-influenza activities. Herein, we described the synthesis of C-4-modified sialic acid derivatives via conjugation with naturally derived pentacyclic triterpenes, which are active ingredients of traditional Chinese medicine, and the evaluation of their in vitro anti-influenza virus activity in MDCK cells. Interestingly, a set of configurational isomers was obtained during the de-O-acetylation reaction of two pentacyclic triterpene–sialic acid conjugates under Zemplén conditions, and a mechanism was proposed. Owing to the attachment of the Neu5Ac2en moiety, all synthesized conjugates displayed lower hydrophobicity than their parent compounds. In comparison with ursane- and lupane-type triterpenes, oleanane-type triterpene-functionalized Neu5Ac2en conjugates were most promising. The insertion of a (1,2,3-triazol-4-yl)-methyl between the amide bond and Neu5Ac2en caused a substantial decrease in activity. Compound 15a exhibited the highest inhibitory activity (IC50 = 8.3 μM) and selectivity index (SI = 22.7). Further studies involving hemagglutination inhibition and neuraminidase inhibition suggested that compound 15a inhibited virus-induced hemagglutination with no effect on the enzymatic activity of neuraminidase, indicating that the antiviral activity appeared to be mediated via interaction with hemagglutinin at the initial stage of viral infection.
 Please wait while we load your content...
Please wait while we load your content...