
A computational framework for distinguishing direct versus indirect interactions in human functional protein–protein interaction networks†
 *a
*a
Abstract
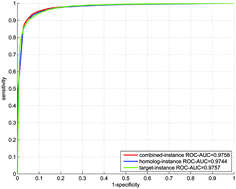
Recognition of indirect interactions is instrumental to in silico reconstruction of signaling pathways and sheds light on the exploration of unknown physical paths between two indirectly interacting genes. However, very limited computational methods have explicitly exploited the indirect interactions with experimental evidence thus far. In this work, we attempt to distinguish direct versus indirect interactions in human functional protein–protein interaction (PPI) networks via a predictive l2-regularized logistic regression model built on the experimental data. The l2-regularized logistic regression method is adopted to counteract the potential homolog noise and reduce the computational complexity on large training data. Computational results show that the proposed model demonstrates promising performance even though the training data are highly skewed. From the 304 799 PPIs that are curated in several databases, the proposed method detects 23 131 indirect interactions, most of which have been verified by the breadth-first graph search algorithm to find dozens of physical paths between the interacting partners. Pathway enrichment analysis shows that most of the physical paths can be mapped onto more than one human signaling pathway, indicating that there do exist a series of biochemical signals between the two indirectly interacting genes. The interactome-scale computational results promise to provide useful cues to the following applications: (1) exploration of unknown physical PPIs or physical paths between two indirectly interacting genes; (2) amending or extending the existing signaling pathways; (3) recognition of the physical PPIs for druggable target discovery.
 Please wait while we load your content...
Please wait while we load your content...