
Towards identifying the active sites on RuO2(110) in catalyzing oxygen evolution†
 a
Manuel J.
Kolb,
‡b
Anders Filsøe
Pedersen,
d
Zhenxing
Feng,
h
Tejs
Vegge,
c
Ib
Chorkendorff,
d
Yang
Shao-Horn
*abg
a
Manuel J.
Kolb,
‡b
Anders Filsøe
Pedersen,
d
Zhenxing
Feng,
h
Tejs
Vegge,
c
Ib
Chorkendorff,
d
Yang
Shao-Horn
*abg
Abstract
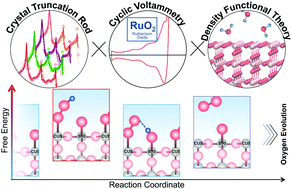
While the surface atomic structure of RuO2 has been well studied in ultra high vacuum, much less is known about the interaction between water and RuO2 in aqueous solution. In this work, in situ surface X-ray scattering measurements combined with density functional theory (DFT) were used to determine the surface structural changes on single-crystal RuO2(110) as a function of potential in acidic electrolyte. The redox peaks at 0.7, 1.1 and 1.4 V vs. reversible hydrogen electrode (RHE) could be attributed to surface transitions associated with the successive deprotonation of –H2O on the coordinatively unsaturated Ru sites (CUS) and hydrogen adsorbed to the bridging oxygen sites. At potentials relevant to the oxygen evolution reaction (OER), an –OO species on the Ru CUS sites was detected, which was stabilized by a neighboring –OH group on the Ru CUS or bridge site. Combining potential-dependent surface structures with their energetics from DFT led to a new OER pathway, where the deprotonation of the –OH group used to stabilize –OO was found to be rate-limiting.
- This article is part of the themed collection: 2017 Energy and Environmental Science HOT articles
 Please wait while we load your content...
Please wait while we load your content...

