
Decamethyltitanocene hydride intermediates in the hydrogenation of the corresponding titanocene-(η2-ethene) or (η2-alkyne) complexes and the effects of bulkier auxiliary ligands†
 *a
*a
Abstract
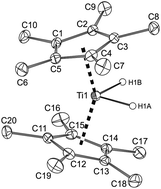
1H NMR studies of reactions of titanocene [Cp*2Ti] (Cp* = η5-C5Me5) and its derivatives [Cp*(η5:η1-C5Me4CH2)TiMe] and [Cp*2Ti(η2-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)] with excess dihydrogen at room temperature and pressures lower than 1 bar revealed the formation of dihydride [Cp*2TiH2] (1) and the concurrent liberation of either methane or ethane, depending on the organometallic reactant. The subsequent slow decay of 1 yielding [Cp*2TiH] (2) was mediated by titanocene formed in situ and controlled by hydrogen pressure. The crystalline products obtained by evaporating a hexane solution of fresh [Cp*2Ti] in the presence of hydrogen contained crystals having either two independent molecules of 1 in the asymmetric part of the unit cell or cocrystals consisting of 1 and [Cp*2Ti] in a 2 : 1 ratio. Hydrogenation of alkyne complexes [Cp*2Ti(η2-R1C
CH2)] with excess dihydrogen at room temperature and pressures lower than 1 bar revealed the formation of dihydride [Cp*2TiH2] (1) and the concurrent liberation of either methane or ethane, depending on the organometallic reactant. The subsequent slow decay of 1 yielding [Cp*2TiH] (2) was mediated by titanocene formed in situ and controlled by hydrogen pressure. The crystalline products obtained by evaporating a hexane solution of fresh [Cp*2Ti] in the presence of hydrogen contained crystals having either two independent molecules of 1 in the asymmetric part of the unit cell or cocrystals consisting of 1 and [Cp*2Ti] in a 2 : 1 ratio. Hydrogenation of alkyne complexes [Cp*2Ti(η2-R1C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR2)] (R1 = R2 = Me or Et) performed at room temperature afforded alkanes R1CH2CH2R2, and after removing hydrogen, 2 was formed in quantitative yields. For alkyne complexes containing bulkier substituent(s) R1 = Me or Ph, R2 = SiMe3, and R1 = R2 = Ph or SiMe3, successful hydrogenation required the application of increased temperatures (70–80 °C) and prolonged reaction times, in particular for bis(trimethylsilyl)acetylene. Under these conditions, no transient 1 was detected during the formation of 2. The bulkier auxiliary ligands η5-C5Me4tBu and η5-C5Me4SiMe3 did not hinder the addition of dihydrogen to the corresponding titanocenes [(η5-C5Me4tBu)2Ti] and [(η5-C5Me4SiMe3)2Ti] yielding [(η5-C5Me4tBu)2TiH2] (3) and [(η5-C5Me4SiMe3)2TiH2] (4), respectively. In contrast to 1, the dihydride 4 did not decay with the formation of titanocene monohydride, but dissociated to titanocene upon dihydrogen removal. The monohydrides [(η5-C5Me4tBu)2TiH] (5) and [(η5-C5Me4SiMe3)2TiH] (6) were obtained by insertion of dihydrogen into the intramolecular titanium-methylene σ-bond in compounds [(η5-C5Me4tBu)(η5:η1-C5Me4CMe2CH2)Ti] and [(η5-C5Me4SiMe3)(η5:η1-C5Me4SiMe2CH2)Ti], respectively. The steric influence of the auxiliary ligands became clear from the nature of the products obtained by reacting 5 and 6 with butadiene. They appeared to be the exclusively σ-bonded η1-but-2-enyl titanocenes (7) and (8), instead of the common π-bonded derivatives formed for the sterically less congested titanocenes, including [Cp*2Ti(η3-(1-methylallyl))] (9). The molecular structure optimized by DFT for compound 1 acquired a distinctly lower total energy than the analogously optimized complex with a coordinated dihydrogen [Cp*2Ti(η2-H2)]. The stabilization energies of binding the hydride ligands to the bent titanocenes were estimated from counterpoise computations; they showed a decrease in the order 1 (−132.70 kJ mol−1), 3 (−121.11 kJ mol−1), and 4 (−112.35 kJ mol−1), in accordance with the more facile dihydrogen dissociation.
CR2)] (R1 = R2 = Me or Et) performed at room temperature afforded alkanes R1CH2CH2R2, and after removing hydrogen, 2 was formed in quantitative yields. For alkyne complexes containing bulkier substituent(s) R1 = Me or Ph, R2 = SiMe3, and R1 = R2 = Ph or SiMe3, successful hydrogenation required the application of increased temperatures (70–80 °C) and prolonged reaction times, in particular for bis(trimethylsilyl)acetylene. Under these conditions, no transient 1 was detected during the formation of 2. The bulkier auxiliary ligands η5-C5Me4tBu and η5-C5Me4SiMe3 did not hinder the addition of dihydrogen to the corresponding titanocenes [(η5-C5Me4tBu)2Ti] and [(η5-C5Me4SiMe3)2Ti] yielding [(η5-C5Me4tBu)2TiH2] (3) and [(η5-C5Me4SiMe3)2TiH2] (4), respectively. In contrast to 1, the dihydride 4 did not decay with the formation of titanocene monohydride, but dissociated to titanocene upon dihydrogen removal. The monohydrides [(η5-C5Me4tBu)2TiH] (5) and [(η5-C5Me4SiMe3)2TiH] (6) were obtained by insertion of dihydrogen into the intramolecular titanium-methylene σ-bond in compounds [(η5-C5Me4tBu)(η5:η1-C5Me4CMe2CH2)Ti] and [(η5-C5Me4SiMe3)(η5:η1-C5Me4SiMe2CH2)Ti], respectively. The steric influence of the auxiliary ligands became clear from the nature of the products obtained by reacting 5 and 6 with butadiene. They appeared to be the exclusively σ-bonded η1-but-2-enyl titanocenes (7) and (8), instead of the common π-bonded derivatives formed for the sterically less congested titanocenes, including [Cp*2Ti(η3-(1-methylallyl))] (9). The molecular structure optimized by DFT for compound 1 acquired a distinctly lower total energy than the analogously optimized complex with a coordinated dihydrogen [Cp*2Ti(η2-H2)]. The stabilization energies of binding the hydride ligands to the bent titanocenes were estimated from counterpoise computations; they showed a decrease in the order 1 (−132.70 kJ mol−1), 3 (−121.11 kJ mol−1), and 4 (−112.35 kJ mol−1), in accordance with the more facile dihydrogen dissociation.
 Please wait while we load your content...
Please wait while we load your content...

