
Computational study of An–X bonding (An = Th, U; X = p-block-based ligands) in pyrrolic macrocycle-supported complexes from the quantum theory of atoms in molecules and bond energy decomposition analysis†

Abstract
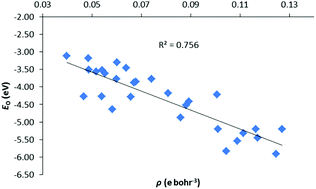
A systematic computational study of organoactinide complexes of the form [LAnX]n+ has been carried out using density functional theory, the quantum theory of atoms in molecules (QTAIM) and Ziegler-Rauk energy decomposition analysis (EDA) methods. The systems studied feature L = trans-calix[2]benzene[2]pyrrolide, An = Th(IV), Th(III), U(III) and X = BH4, BO2C2H4, Me, N(SiH3)2, OPh, CH3, NH2, OH, F, SiH3, PH2, SH, Cl, CH2Ph, NHPh, OPh, SiH2Ph, PHPh2, SPh, CPh3, NPh2, OPh, SiPh3 PPh2, SPh. The PBE0 hybrid functional proved most suitable for geometry optimisations based on comparisons with available experimental data. An–X bond critical point electron densities, energy densities and An–X delocalisation indices, calculated with the PBE functional at the PBE0 geometries, are correlated with An–X bond energies, enthalpies and with the terms in the EDA. Good correlations are found between energies and QTAIM metrics, particularly for the orbital interaction term, provided the X ligand is part of an isoelectronic series and the number of open shell electrons is low (i.e. for the present Th(IV) and Th(III) systems).
 Please wait while we load your content...
Please wait while we load your content...

