
A phenoxo-bridged dicopper(ii) complex as a model for phosphatase activity: mechanistic insights from a combined experimental and computational study†‡

Abstract
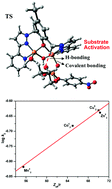
A μ-phenoxo-bis(μ2-1,3-acetato)-bridged dicopper(II) complex [CuII2(L1)(μ-O2CMe)2][NO3] (1) has been synthesized from the perspective of modeling phosphodiesterase activity. Structural characterization was done initially with 1·3Et2O (vapour diffusion of Et2O into MeOH solution of 1; poor crystal quality) and finally with its perchlorate salt [CuII2(L1)(μ-O2CMe)2][ClO4]·1.375MeCN·0.25H2O, crystallized from vapour diffusion of n-pentane into a MeCN–MeOH mixture (comparatively better crystal quality). An asymmetric unit of such a crystal contains two independent molecules of compositions [CuII2(L1)(μ-O2CMe)2][ClO4] and [CuII2(L1)(μ-O2CMe)2(MeCN)][ClO4] (coordinated MeCN with 0.75 occupancy), and two molecules of MeCN and H2O (each H2O molecule with 0.25 occupancy) as the solvent of crystallization. These two cations, each having five-coordinate (μ-phenoxo)bis(μ-acetato)-bridged CuII ions, differ by only the coordination environment of only one CuII ion, which has a weakly coordinated acetonitrile molecule in its sixth position. Temperature-dependent magnetic studies on 1 reveal that the copper(II) centres are antiferromagnetically coupled with the exchange-coupling constant J = −124(1) cm−1. Theoretically calculated J = −126.51 cm−1, employing a broken-symmetry DFT approach, is in excellent agreement with the experimental value. The dicopper(II) complex has been found to be catalytically efficient in the hydrolysis of 2-hydroxypropyl-p-nitrophenylphosphate (HPNP). Detailed kinetic experiments and solution studies (potentiometry, species distribution and ESI-MS) were performed to elucidate the reaction mechanism. DFT calculations were performed to discriminate between different possible mechanistic pathways. The free-energy barrier for HPNP hydrolysis catalyzed by 1 is comparable to that obtained from the experimentally-determined value. The involvement of non-covalent (hydrogen-bonding) interaction has also been probed by DFT calculations. The activity of 1 is found to be the highest, compared to the structurally-characterized MnII2, CoII2, NiII2 and ZnII2 complexes of L1(−) reported earlier, under identical experimental conditions, in which each metal centre is six-coordinate.
 Please wait while we load your content...
Please wait while we load your content...

