
Infrared spectroscopy of hydrated polycyclic aromatic hydrocarbon cations: naphthalene+–water†
 *a
*a
Abstract
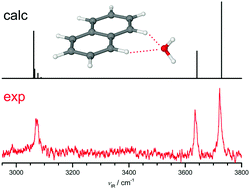
Polycyclic aromatic hydrocarbons (PAHs) are suggested to occur in interstellar media and ice grains. It is important to characterize hydrated PAHs and their cations to explore their stability in interstellar and biological media. Herein, the infrared photodissociation (IRPD) spectrum of the naphthalene+–H2O radical cation (Np+–H2O) recorded in the O–H and C–H stretch range is analysed by dispersion-corrected density functional theory calculations at the B3LYP-D3/aug-cc-pVTZ level to determine its structure and intermolecular bonding. Monohydration of Np+ in its 2Au ground electronic state leads to the formation of a bifurcated CH⋯O ionic hydrogen bond (H-bond), in which the lone pairs of H2O bind to two adjacent CH proton donors of the two aromatic rings. The frequency-dependent branching ratios observed for IRPD of cold Np+–H2O–Ar clusters allows the estimation of the dissociation energy of Np+–H2O as D0 ∼ 2800 ± 300 cm−1. The monohydration motif of Np+ differs qualitatively from that of the benzene cation in both structure and binding energy, indicating the strong influence of the multiple aromatic rings on the hydration of PAH+ cations. This difference is rationalized by natural bond orbital analysis of the ionic H-bond motif. Comparison with neutral Np–H2O reveals the large change in structure and bond strength of the hydrated PAHs upon ionization. While neutral Np–H2O is stabilized by weak π H-bonds (OH⋯π, π-stacking), strong cation–dipole forces favour a planar bifurcated CH⋯O ionic H-bond in Np+–H2O.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

