
Vibrational spectroscopic characterization of arylisoquinolines by means of Raman spectroscopy and density functional theory calculations†
 *b
Torsten
Frosch
*acd
*b
Torsten
Frosch
*acd
Abstract
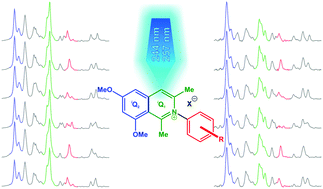
Promising new antimalarial agents were investigated using FT-NIR and deep-UV resonance Raman spectroscopy. The Raman spectra of the seven arylisoquinolines (AIQ) were calculated with the help of density functional theory (DFT). Very good agreement with the experimental data was achieved and a convincing mode assignment was performed with the help of the calculated potential energy distribution (PED). For the non-resonant Raman spectra the most prominent bands were assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching modes of the isoquinoline system. To differentiate between substances with similar structures, deep-UV resonance Raman spectra were recorded. Raman bands in the range between 1250 and 1210 cm−1 were assigned to ν(CC) stretching vibrations in combination with δ(HCC) deformation vibrations of the aryl rests. These vibrations of the aryl part of the molecules were selectively enhanced, which, thus, enabled the differentiation of similar active agents from each other. For λexc = 257 nm excitation, strong ν(CC) vibrations of the isoquinoline (benzo-) part dominate the Raman spectrum in the range between 1685 and 1585 cm−1 and for λexc = 244 nm the Raman signals between 1430 and 1350 cm−1 were enhanced and assigned to ν(CC) of the isoquinoline (pyridino-) system.
C) stretching modes of the isoquinoline system. To differentiate between substances with similar structures, deep-UV resonance Raman spectra were recorded. Raman bands in the range between 1250 and 1210 cm−1 were assigned to ν(CC) stretching vibrations in combination with δ(HCC) deformation vibrations of the aryl rests. These vibrations of the aryl part of the molecules were selectively enhanced, which, thus, enabled the differentiation of similar active agents from each other. For λexc = 257 nm excitation, strong ν(CC) vibrations of the isoquinoline (benzo-) part dominate the Raman spectrum in the range between 1685 and 1585 cm−1 and for λexc = 244 nm the Raman signals between 1430 and 1350 cm−1 were enhanced and assigned to ν(CC) of the isoquinoline (pyridino-) system.
 Please wait while we load your content...
Please wait while we load your content...

