
Surface phase diagram prediction from a minimal number of DFT calculations: redox-active adsorbates on zinc oxide
 *a
and
Jörg
Behler
a
*a
and
Jörg
Behler
a
Abstract
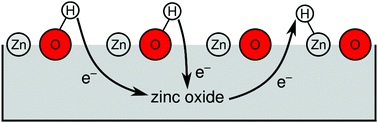
Density functional theory (DFT) is routinely used to calculate the adsorption energies of molecules on solid surfaces, which can be employed to derive surface phase diagrams. Such calculations become computationally expensive if the number of substrate atoms is large, which happens whenever the adsorbate coverage is small. Here, we propose an efficient method for calculating surface phase diagrams for redox-active adsorbates on semiconductors, that we apply to the important example of proton (H+) and hydride (H−) adsorbates on a ZnO surface. We identify the leading cause for the coverage dependence of the adsorption energies to be the filling and depletion of the disperse substrate conduction band. From only four DFT calculations, coupled with an analysis of the substrate electronic band structure and changes in the electrostatic potential within the substrate upon adsorption, we derive a phenomenological model that well describes the coverage-dependent adsorption energies. Moreover, our model allows us to extrapolate to the “infinite” supercell limit, where additional H adsorption leads to an arbitrarily small increase of the surface coverage. With this tool we are able to derive a surface phase diagram containing structures with extremely small H coverages (<0.002 ML), that have so far been unattainable. We expect that such models can be applied to a wide range of semiconductor substrates and redox-active adsorbates.
 Please wait while we load your content...
Please wait while we load your content...

