
Tunable interaction between metal clusters and graphene†

Abstract
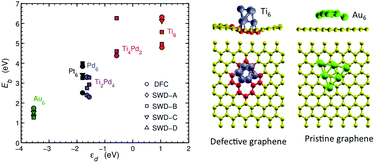
We describe the interaction between small transition metal clusters and graphene using first principles calculations. The coupling is analyzed in terms of different features of the system: binding energy, decomposition into atomic orbitals, the presence of defects on the graphene layer, and both the band and geometrical structures. The binding strength is found to follow the d-band model, which anticipates the binding energies of clusters on graphene layers from the position of the cluster's d-band centers relative to the their highest-occupied and lowest-unoccupied molecular orbital levels. These findings are verified for 6-atom and 13-atom transition metal clusters (Ti, Pd, Pt, and Au) and considering different types of defects. The adhesion of the TM clusters is substantially larger on defective graphene layers than on pristine ones. Buckling of the graphene layer may arise from the presence of defects but it does not necessarily imply strong binding. However, buckling can sometimes offer configurational paths through which the adsorbed cluster is stabilized changing its original shape. Insights into this work offer mechanisms to tailor the electronic properties of the combined nanoparticle–graphene system by changing the size and composition of transition metal clusters.
 Please wait while we load your content...
Please wait while we load your content...

