
Fragmentation of KrN+ clusters after electron impact ionization II. Long-time dynamics simulations of Kr7+ evolution and the role of initial electronic excitation†
 *a
René
Kalus
*bd
*a
René
Kalus
*bd
Abstract
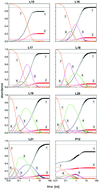
Long time simulations, up to 100 ns, have been performed for the fragmentation of Kr7+ clusters after electron impact ionization. They rely on DIM approaches and hybrid non-adiabatic dynamics combining mean field and decoherence driven either by Tully fewest switches (TFS) algorithm or through electronic amplitude (AMP) calculations. With both methods, for the first time, when the initial electronic excited state belongs to group II correlating to P1/2 atomic ions, the fragmentation ratio in mainly monomer and dimer ions agrees very well with known experimental results. A complex non-adiabatic dynamics is found where initial neutral monomer evaporations due to gradual deexcitation over electronic states of group II are followed by a non-adiabatic transition across a wide energy gap of the spin–orbit origin to electronic states of group I. The resulting excess of kinetic energy causes the final fragmentation of charged intermediate fragments to stable ionic monomers or dimers. Characteristic times of these processes have been estimated. The kinetic energy distribution of the neutral and ionic monomers (the dominating final fragments) has been analyzed in detail. Interestingly they exhibit some signature of the initial excited electronic state which could allow for an experimental identification.
 Please wait while we load your content...
Please wait while we load your content...

