
Unveiling universal trends for the energy level alignment in organic/oxide interfaces
 *a
José
Ortega,
b
*a
José
Ortega,
b
Abstract
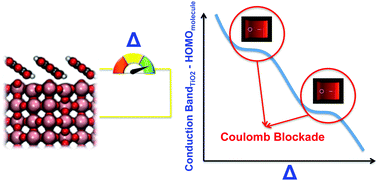
In this perspective we present a comprehensive analysis of the energy level alignment at the interface between an organic monolayer (organic = perylenetetracarboxylic dianhydride, PTCDA, zinc tetraphenylporphyrin, Zn-TPP, and tetracyanoquinodimethane, TCNQ) and a prototypical oxide surface, TiO2(110), looking for universal behaviours. PTCDA shows a physisorbed interaction with TiO2 and a small interface dipole potential with its highest occupied molecular orbital (HOMO) energy level located in the oxide energy gap and the lowest occupied molecular orbital (LUMO) energy level located above the oxide conduction band minimum, EC. We analyse how the interface barrier depends on an external bias potential between the organic layer and the oxide surface, Δ, and find for this interface that the screening parameter S = d|(EC − HOMO)|dΔ is close to 1. In the second case, the Zn-TPP monolayer shows a moderate chemisorbed interaction with some charge transfer from the molecule to the oxide and a significant interface dipole potential, in such a way that S decreases to around 0.8. In the TCNQ/TiO2(110) case, the TCNQ molecules present a strong chemical interaction with the oxide; the LUMO energy level is located in the oxide energy gap in such a way that one electron is transferred from the oxide to the organic molecule; we also find that in this case S ≈ 0.5. All these cases can be integrated within a universal behaviour when (EC − HOMO) is calculated as a function of Δ; that function presents a zig-zag curve with a central part having an S-slope, and two plateaus associated with either the LUMO or the HOMO energy levels crossing the oxide Fermi level. In these plateaus, a Coulomb blockade regime arises and a pace charge layer develops in the oxide surface.
- This article is part of the themed collections: PCCP Perspectives and 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

