
Stepwise deprotonation of sumanene: electronic structures, energetics and aromaticity alterations†
 b
and
Andrey Yu.
Rogachev
*a
b
and
Andrey Yu.
Rogachev
*a
Abstract
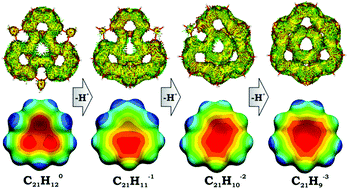
The first comprehensive theoretical investigation of structural, energetic, and electronic changes in a sumanene skeleton, C21H12, upon a step-wise deprotonation process is performed. This study is complemented by a detailed consideration of aromaticity in target bowl-shaped systems, including neutral sumanene and its three deprotonated anions, namely C21H111−, C21H102−, and C21H93−. In order to obtain the most reliable and method-independent characteristics, a set of aromatic descriptors of different nature has been applied. It included structure-based HOMA, topological descriptors PDI and FLU, as well as magnetic NICS and ACID. The calculation results reveal that the neutral sumanene can be best described as mechanically bent triphenylene, in which π-conjugation is mostly localized over three peripheral 6-membered rings. Sequential deprotonation changed the system from the localized mono-anionic to semi-localized di-anionic, and eventually to the fully delocalized tri-anionic sumanenyl species. Structural changes, namely, bond equalization upon the deprotonation process, are in excellent agreement with alterations observed in electronic structures and aromaticity. Deprotonation results in a significant reduction of the barrier for a bowl-to-bowl transition only in the tri-anionic sumanenyl system, whereas the first and the second deprotonation steps show no notable effect. This clearly indicates that only complete aromatization of the sumanene core in C21H93− leads to a substantial increase of bowl flexibility.
 Please wait while we load your content...
Please wait while we load your content...

