
The structural and electronic properties of reduced amorphous titania†
 *a
*a
Abstract
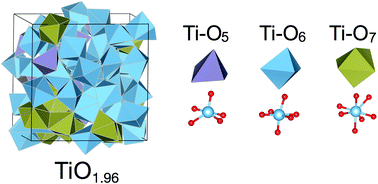
Crystalline titania has been extensively studied using experimental and theoretical tools. Amorphous titania, however, has received less attention in the literature, despite its importance for a number of applications, such as photocatalysis, batteries, and electronic devices. In this work we modeled amorphous titania using a combination of molecular dynamics and density functional theory with several stoichiometries (TiOx, 2 ≥ x ≥ 1.75). Our results show that oxygen atom removal from amorphous titania is much easier than from crystalline titania, indicating that reduced amorphous structures are likely common. Ti atoms in amorphous titania exhibit a distribution of coordination numbers (five to seven), but the average coordination number of oxygen increases upon reduction. We also identified that gap states arise in substoichiometric titania due to the formation of Ti3+ centers. Such gap states are highly localized and randomly distributed across different Ti atoms, although we do observe a slight preference for electron localization on seven-coordinated Ti atoms. We observe that band gaps increase with reduction of amorphous titania. We also analyzed a proposed hole hopping mechanism involving oxygen vacancies by calculating hole hopping distances. We found that such distances are large except in very reduced states, indicating likely slow hole diffusion through an oxygen vacancy mechanism. Our work is the first of its kind to thoroughly characterize the structural and electronic properties of amorphous titania in reduced states.
 Please wait while we load your content...
Please wait while we load your content...

