
Multiscale simulations for understanding the evolution and mechanism of hierarchical peptide self-assembly
 *abc
*abc
Abstract
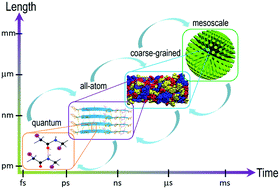
Hierarchical self-assembly, abundant in biological systems, has been explored as an effective bottom-up method to fabricate highly ordered functional superstructures from elemental building units. Biomolecules, especially short peptides consisting of several amino acids, are a type of elegant building blocks due to their advantages of structural, mechanical, and functional diversity as well as high biocompatibility and biodegradability. The hierarchical self-assembly of peptides is a spontaneous process spanning multiple time and length scales under certain thermodynamics and kinetics conditions. Therefore, understanding the mechanisms of dynamic processes is crucial to directing the construction of complicated biomimetic systems with multiple functionalities. Multiscale molecular simulations that combine and systematically link several hierarchies can provide insights into the evolution and dynamics of hierarchical self-assembly from the molecular level to the mesoscale. Herein, we provided an overview of the simulation hierarchies in the general field of peptide self-assembly modeling. In particular, we highlighted multiscale simulations for unraveling the mechanisms underlying the dynamic self-assembly process with an emphasis on weak intermolecular interactions in the process stages and the energies of different molecular alignments as well as the role of thermodynamic and kinetic factors at the microscopic level.
- This article is part of the themed collections: PCCP Perspectives, 2017 PCCP HOT Articles and Surface chemistry and interface science
 Please wait while we load your content...
Please wait while we load your content...

