
Cation solvation with quantum chemical effects modeled by a size-consistent multi-partitioning quantum mechanics/molecular mechanics method†
 *ab
Maximilian
Kubillus,
c
Tomáš
Kubař,
c
Boris
Mizaikoff
d
and
Hiroshi
Ishikita
ab
*ab
Maximilian
Kubillus,
c
Tomáš
Kubař,
c
Boris
Mizaikoff
d
and
Hiroshi
Ishikita
ab
Abstract
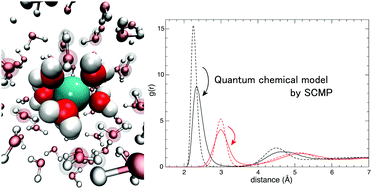
In the condensed phase, quantum chemical properties such as many-body effects and intermolecular charge fluctuations are critical determinants of the solvation structure and dynamics. Thus, a quantum mechanical (QM) molecular description is required for both solute and solvent to incorporate these properties. However, it is challenging to conduct molecular dynamics (MD) simulations for condensed systems of sufficient scale when adapting QM potentials. To overcome this problem, we recently developed the size-consistent multi-partitioning (SCMP) quantum mechanics/molecular mechanics (QM/MM) method and realized stable and accurate MD simulations, using the QM potential to a benchmark system. In the present study, as the first application of the SCMP method, we have investigated the structures and dynamics of Na+, K+, and Ca2+ solutions based on nanosecond-scale sampling, a sampling 100-times longer than that of conventional QM-based samplings. Furthermore, we have evaluated two dynamic properties, the diffusion coefficient and difference spectra, with high statistical certainty. Furthermore the calculation of these properties has not previously been possible within the conventional QM/MM framework. Based on our analysis, we have quantitatively evaluated the quantum chemical solvation effects, which show distinct differences between the cations.
 Please wait while we load your content...
Please wait while we load your content...

