
Path integral-GC-AdResS simulation of a large hydrophobic solute in water: a tool to investigate the interplay between local microscopic structures and quantum delocalization of atoms in space

Abstract
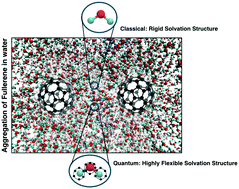
We perform large scale quantum (path integral) molecular dynamics simulations of a C60-like molecule in water. The path integral (PI) technique allows for the description of the delocalization of atoms in space and of its consequences on the structure and dynamics of the hydrogen bonding network around the solute. We then employ the adaptive resolution method GC-AdResS, which unambiguously defines the essential (necessary) degrees of freedom required for a certain property, to analyze the locality of the water structure around the solute. We show that the feature of locality is independent of the use of a quantum or classical model of water. However the water structure around the solute obtained from classical simulations is more ordered and rigid than the structure found in quantum simulations. With this study we mainly intend to show that GC-AdResS, besides its computational efficiency, can be used as a powerful tool of multiscale analysis; this capability, in turn, can be used to speculate about processes at larger scales. We make an example, whose current validity is restricted to the molecular models specifically used, regarding the possible role of quantum effects in the aggregation of fullerene-like molecules in water.
 Please wait while we load your content...
Please wait while we load your content...

