
Modeling the absorption spectrum of the permanganate ion in vacuum and in aqueous solution
 *a
and
*a
and
Abstract
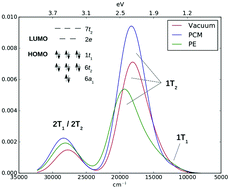
The absorption spectrum of the MnO4− ion has been a test-bed for quantum-chemical methods over the last decades. Its correct description requires highly-correlated multiconfigurational methods, which are incompatible with the inclusion of finite-temperature and solvent effects due to their high computational demands. Therefore, implicit solvent models are usually employed. Here we show that implicit solvent models are not sufficiently accurate to model the solvent shift of MnO4−, and we analyze the origins of their failure. We obtain the correct solvent shift for MnO4− in aqueous solution by employing the polarizable embedding (PE) model combined with a range-separated complete active space short-range density functional theory method (CAS-srDFT). Finite-temperature effects are taken into account by averaging over structures obtained from ab initio molecular dynamics simulations. The explicit treatment of finite-temperature and solvent effects facilitates the interpretation of the bands in the low-energy region of the MnO4− absorption spectrum, whose assignment has been elusive.
 Please wait while we load your content...
Please wait while we load your content...

