
The rotational dynamics of H2 adsorbed in covalent organic frameworks†
 c
Juergen
Eckert
*ad
and
c
Juergen
Eckert
*ad
and
Abstract
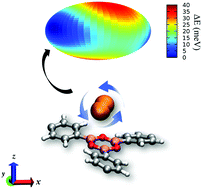
A combined inelastic neutron scattering (INS) and theoretical study was carried out on H2 adsorbed in two covalent organic framework (COF) materials: COF-1 and COF-102. These COFs are synthesized from self-condensation reactions of 1,4-benzenediboronic acid (BDBA) and tetra(4-(dihydroxy)borylphenyl)methane (TBPM) molecules, respectively. Molecular simulations of H2 adsorption in COF-1 revealed that the H2 molecules occupy the region between two eclipsed layers of the COF. The most favorable H2 binding site in COF-1 is located between two B3O3 clusters of the eclipsed layers. Two distinct H2 binding sites were identified in COF-102 from the simulations: the B3O3 clusters and the phenyl rings of the tetraphenylmethyl units. Two-dimensional quantum rotation calculations for H2 adsorbed at the considered sites in both COFs resulted in rotational transitions that are in good agreement with those that appear in the corresponding INS spectra. Such calculations were important for interpreting the INS spectra in these materials. Calculation of the rotational potential energy surface for H2 bound at the most favorable adsorption site in COF-1 and COF-102 revealed unusually high rotational barriers that are attributed to the nature of the B3O3 rings. The values for these barriers to rotation are greater than or comparable to those observed in some metal–organic frameworks (MOFs) that possess open-metal sites. This study demonstrates the power of using INS experiments in conjunction with theoretical calculations to gain valuable insights into the nature of the binding sites and, for the first time, the rotational dynamics of H2 adsorbed in COFs.
 Please wait while we load your content...
Please wait while we load your content...

