
A comparative study of surface energies and water adsorption on Ce-bastnäsite, La-bastnäsite, and calcite via density functional theory and water adsorption calorimetry†‡

Abstract
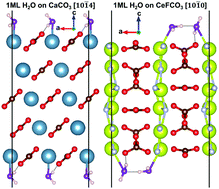
Bastnäsite, a fluoro-carbonate mineral, is the single largest mineral source of light rare earth elements (REE), La, Ce and Nd. Enhancing the efficiency of separation of the mineral from gangue through froth flotation is the first step towards meeting an ever increasing demand for REE. To design and evaluate collector molecules that selectively bind to bastnäsite, a fundamental understanding of the structure and surface properties of bastnäsite is essential. In our earlier work (J. Phys. Chem. C, 2016, 120, 16767), we carried out an extensive study of the structure, surface stability and water adsorption energies of La-bastnäsite. In this work, we make a comparative study of the surface properties of Ce-bastnäsite, La-bastnäsite, and calcite using a combination of density functional theory (DFT) and water adsorption calorimetry. Spin polarized DFT+U calculations show that the exchange interaction between the electrons in Ce 4f orbitals is negligible and that these orbitals do not participate in bonding with the oxygen atom of the adsorbed water molecule. In agreement with calorimetry, DFT calculations predict larger surface energies and stronger water adsorption energies on Ce-bastnäsite than on La-bastnäsite. The order of stabilities for stoichiometric surfaces is as follows: [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] > [101] > [102] > [0001] > [11
0] > [101] > [102] > [0001] > [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 2] > [104] and the most favorable adsorption sites for water molecules are the same as for La-bastnäsite. In agreement with water adsorption calorimetry, at low coverage water molecules are strongly stabilized via coordination to the surface Ce3+ ions, whereas at higher coverage they are adsorbed less strongly via hydrogen bonding interaction with the surface anions. Due to similar water adsorption energies on bastnäsite [101] and calcite [104] surfaces, the design of collector molecules that selectively bind to bastnäsite over calcite must exploit the structural differences in the predominantly exposed facets of these minerals.
2] > [104] and the most favorable adsorption sites for water molecules are the same as for La-bastnäsite. In agreement with water adsorption calorimetry, at low coverage water molecules are strongly stabilized via coordination to the surface Ce3+ ions, whereas at higher coverage they are adsorbed less strongly via hydrogen bonding interaction with the surface anions. Due to similar water adsorption energies on bastnäsite [101] and calcite [104] surfaces, the design of collector molecules that selectively bind to bastnäsite over calcite must exploit the structural differences in the predominantly exposed facets of these minerals.
 Please wait while we load your content...
Please wait while we load your content...

