
The effect of substituents on triply bonded boron![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) antimony molecules: a theoretical approach†
antimony molecules: a theoretical approach†
 *ab
*ab
Abstract
Three (M06-2X/Def2-TZVP, B3PW91/Def2-TZVP and B3LYP/LANL2DZ+dp) levels of theory are used to study the effect of substituents on the potential energy surfaces of RBSbR (R = F, OH, H, CH3, SiH3, SiMe(SitBu3)2, SiiPrDis2 and NHC). The theoretical results demonstrate that the triply bonded RBSbR molecules favor a bent geometry: that is, ∠R–B–Sb ≈ 180° and ∠B–Sb–R ≈ 120°. Regardless of the type of substituents that are attached to the RBSbR compounds, theoretical evidence strongly indicates that their BSb triple bonds have a donor–acceptor nature and are proven to be very weak. Two valence bond models clarify the bonding characters of the BSb triple bond. For RBSbR molecules that feature small substituents, the triple bond is represented as 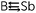 . For RBSbR molecules that feature large substituents, the triple bond is represented as
. For RBSbR molecules that feature large substituents, the triple bond is represented as  . Most importantly, this theoretical study predicts that only bulkier substituents significantly stabilize the triply bonded RBSbR molecules, from the kinetic viewpoint.
. Most importantly, this theoretical study predicts that only bulkier substituents significantly stabilize the triply bonded RBSbR molecules, from the kinetic viewpoint.
![Graphical abstract: The effect of substituents on triply bonded boron [[triple bond, length as m-dash]] antimony molecules: a theoretical approach](/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C7CP00421D&imageInfo.ImageIdentifier.Year=2017)
 Please wait while we load your content...
Please wait while we load your content...
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) antimony molecules: a theoretical approach
antimony molecules: a theoretical approach

