
Molecular dynamics simulations of phosphonic acid–aluminum oxide self-organization and their evolution into ordered monolayers†
 a
a
Abstract
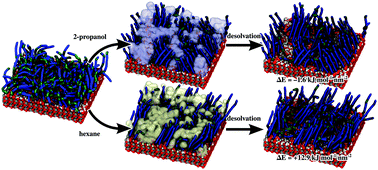
We outline an unprejudiced molecular dynamics simulation approach to study the mechanisms of self-organization encompassing the evolution of surfactant–surface interactions to the growth of self-assembled monolayers (SAMs). Therein, the time-length scale problem is tackled by combining an efficient docking-type procedure for implementing surfactant-by-surfactant association with detailed molecular simulations to explore structural relaxation. For this, nanosecond-scale molecular dynamics simulations unravel ordering processes during the gradual assembly of the monolayer. Along this line, different packing motifs of octadecyl phosphonic acid (ODPA) on the (0001) surface of α-alumina and implications for the final density and ordering of the resulting monolayers are elucidated. Moreover, the role of the solvent is discriminated by comparing SAM formation in 2-propanol, hexane and in a vacuum.
 Please wait while we load your content...
Please wait while we load your content...

