
A kinetic model of water adsorption, clustering and dissociation on the Fe3S4{001} surface†
 *a
and
*a
and
Abstract
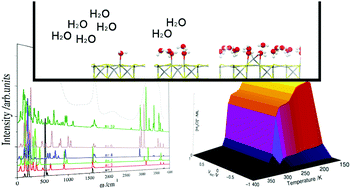
The interaction of water with catalyst surfaces is a common process which requires investigation. Here, we have employed density functional theory calculations to investigate the adsorption of up to ten water molecules on the {001} surface of greigite (Fe3S4), which owing to its redox properties, is of increasing interest as a catalyst, e.g. in electro-catalysis. We have systematically analyzed and characterized the modes of water adsorption on the surface, where we have considered both molecular and dissociative adsorption processes. The calculations show that molecular adsorption is the predominant state on these surfaces, from both a thermodynamic and kinetic point of view. We have explored the molecular dispersion on the surface under different coverages and found that the orientation of the molecule, and therefore the surface dipole, depends on the number of adsorbed molecules. The interactions between the water molecules become stronger with an increasing number of water molecules, following an exponential decay which tends to the interaction energy found in bulk water. We have also shown the evolution of the infra-red signals as a function of water coverage relating to the H-bond networks formed on the surface. Next we have included these results in a classical micro-kinetic model, which introduced the effects of temperature in the simulations, thus helping us to derive the water cluster size on the greigite surface as a function of the initial conditions of pressure, temperature and external potential. The kinetic model concluded that water molecules agglomerate in clusters instead of wetting the surface, which agrees with the low hydrophilicity of Fe3S4. Clusters consisting of four water molecules was shown to be the most stable cluster under a wide range of temperatures and external potential.
 Please wait while we load your content...
Please wait while we load your content...

