
Elucidating the 3D structures of Al(iii)–Aβ complexes: a template free strategy based on the pre-organization hypothesis†

Abstract
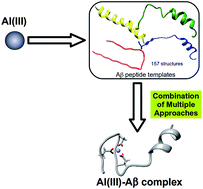
Senile plaques are extracellular deposits found in patients with Alzheimer’s Disease (AD) and are mainly formed by insoluble fibrils of β-amyloid (Aβ) peptides. The mechanistic details about how AD develops are not fully understood yet, but metals such as Cu, Zn, or Fe are proposed to have a non-innocent role. Many studies have also linked the non biological metal aluminum with AD, a species whose concentration in the environment and food has been constantly increasing since the industrial revolution. Gaining a molecular picture of how Al(III) interacts with an Aβ peptide is of fundamental interest to improve understanding of the many variables in the evolution of AD. So far, no consensus has been reached on how this metal interacts with Aβ, partially due to the experimental complexity of detecting and quantifying the resulting Al(III)–Aβ complexes. Computational chemistry arises as a powerful alternative to investigate how Al(III) can interact with Aβ peptides, as suitable strategies could shed light on the metal–peptide description at the molecular level. However, the absence of any reliable template that could be used for the modeling of the metallopeptide structure makes computational insight extremely difficult. Here, we present a novel strategy to generate accurate 3D models of the Al(III)–Aβ complexes, which still circumvents first principles simulations of metal binding to peptides of Aβ. The key to this approach lies in the identification of experimental structures of the isolated peptide that are favourably pre-organized for the binding of a given metal in configurations of the first coordination sphere that were previously identified as the most stable with amino acid models. This approach solves the problem of the absence of clear structural templates for novel metallopeptide constructs. The posterior refinement of the structures via QM/MM and MD calculations allows us to provide, for the first time, physically sound models for Al(III)–Aβ complexes with a 1 : 1 stoichiometry, where up to three carboxylic groups are involved in the metal binding, with a clear preference towards Glu3, Asp7, and Glu11.
 Please wait while we load your content...
Please wait while we load your content...

