
A thiocyanopalladation/carbocyclization transformation identified through enzymatic screening: stereocontrolled tandem C–SCN and C–C bond formation†

Abstract
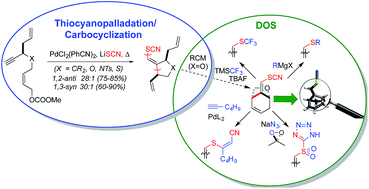
Herein we describe a formal thiocyanopalladation/carbocyclization transformation and its parametrization and optimization using a new elevated temperature plate-based version of our visual colorimetric enzymatic screening method for reaction discovery. The carbocyclization step leads to C–SCN bond formation in tandem with C–C bond construction and is highly stereoselective, showing nearly absolute 1,2-anti-stereoinduction (5 examples) for substrates bearing allylic substitution, and nearly absolute 1,3-syn-stereoinduction (16 examples) for substrates bearing propargylic substitution. Based upon these high levels of stereoinduction, the dependence of the 1,2-stereoinduction upon cyclization substrate geometry, and the generally high preference for the transoid vinyl thiocyanate alkene geometry, a mechanistic model is proposed, involving (i) Pd(II)-enyne coordination, (ii) thiocyanopalladation, (iii) migratory insertion and (iv) β-elimination. Examples of transition metal-mediated C–SCN bond formation that proceed smoothly on unactivated substrates and allow for preservation of the SCN moiety are lacking. Yet, the thiocyanate functionality is of great value for biophysical chemistry (vibrational Stark effect) and medicinal chemistry (S,N-heterocycle construction). The title transformation accommodates C-, O-, N- and S-bridged substrates (6 examples), thereby providing the corresponding carbocyclic or heterocyclic scaffolds. The reaction is also shown to be compatible with a significant range of substituents, varying in steric and electronic demand, including a wide range of substituted aromatics, fused bicyclic and heterocyclic systems, and even biaryl systems. Combination of this new transformation with asymmetric allylation and Grubbs ring-closing metathesis provides for a streamlined enantio- and diastereoselective entry into the oxabicyclo[3.2.1]octyl core of the natural products massarilactone and annuionone A, as also evidenced by low temperature X-ray crystal structure determination. Utilizing this bicyclic scaffold, we demonstrate the versatility of the thiocyanate moiety for structural diversification post-cyclization. Thus, the bridging vinyl thiocyanate moiety is smoothly elaborated into a range of derivative functionalities utilizing transformations that cleave the S–CN bond, add the elements of RS-CN across a π-system and exploit the SCN moiety as a cycloaddition partner (7 diverse examples). Among the new functionalities thereby generated are thiotetrazole and sulfonyl tetrazole heterocycles that serve as carboxylate and phosphate surrogates, respectively, highlighting the potential of this approach for future applications in medicinal chemistry or chemical biology.
 Please wait while we load your content...
Please wait while we load your content...

