
Examining the role of protein structural dynamics in drug resistance in Mycobacterium tuberculosis†
 ‡a
Anthony W.
Parker,
d
David
Robinson,
e
Jonathan D.
Hirst,
*b
Paul A.
Hoskisson
*c
and
Neil T.
Hunt
*a
‡a
Anthony W.
Parker,
d
David
Robinson,
e
Jonathan D.
Hirst,
*b
Paul A.
Hoskisson
*c
and
Neil T.
Hunt
*a
Abstract
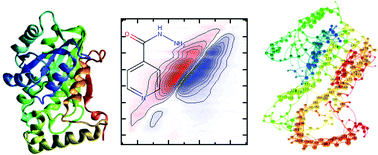
Antimicrobial resistance represents a growing global health problem. The emergence of novel resistance mechanisms necessitates the development of alternative approaches to investigate the molecular fundamentals of resistance, leading ultimately to new strategies for counteracting them. To gain deeper insight into antibiotic–target interactions, the binding of the frontline anti-tuberculosis drug isoniazid (INH) to a target enzyme, InhA, from Mycobacterium tuberculosis was studied using ultrafast two-dimensional infrared (2D-IR) spectroscopy and molecular simulations. Comparing wild-type InhA with a series of single point mutations, it was found that binding of the INH–NAD inhibitor to susceptible forms of the enzyme increased the vibrational coupling between residues located in the Rossmann fold co-factor binding site of InhA and suppressed dynamic fluctuations of the enzyme structure. The effect correlated with biochemical assay data, being reduced in the INH-resistant S94A mutant and absent in the biochemically-inactive P193A control. Molecular dynamics simulations and calculations of inter–residue couplings indicate that the changes in coupling and dynamics are not localised to the co-factor binding site, but permeate much of the protein. We thus propose that the resistant S94A mutation circumvents subtle changes in global structural dynamics caused by INH upon binding to the wild-type enzyme that may impact upon the formation of important protein–protein complexes in the fatty acid synthase pathway of M. tuberculosis.
 Please wait while we load your content...
Please wait while we load your content...

