
Biomolecular conformational changes and ligand binding: from kinetics to thermodynamics†

Abstract
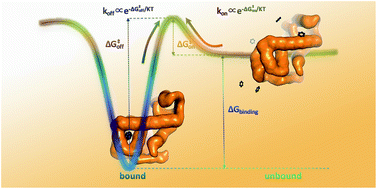
The behaviour of biomolecular systems is governed by their thermodynamic and kinetic properties. It is thus important to be able to calculate, for example, both the affinity and rate of binding and dissociation of a protein–ligand complex, or the populations and exchange rates between distinct conformational states. Because these are typically rare events, calculating these properties from long molecular dynamics simulations remains extremely difficult. Instead, one often adopts a divide-and-conquer strategy in which equilibrium free-energy differences and the fastest state-to-state transition (e.g. ligand association or minor-to-major state conversion) are combined to estimate the slow rate (e.g. ligand dissociation) using a two-state assumption. Here we instead address these problems by using a previously developed method to calculate both the forward and backward rates directly from simulations. We then estimate the thermodynamics from the rates, and validate these values by independent means. We applied the approach to three systems of increasing complexity, including the association and dissociation of benzene to a fully buried cavity inside the L99A mutant variant of T4 lysozyme. In particular, we were able to determine both millisecond association and dissociation rates, and the affinity, of the protein–ligand system by directly observing dozens of rare events in atomic detail. Our approach both sheds light on the precision of methods for calculating kinetics and further provides a generally useful test for the internal consistency of kinetics and thermodynamics. We also expect our route to be useful for obtaining both the kinetics and thermodynamics at the same time in more challenging cases.
 Please wait while we load your content...
Please wait while we load your content...

