
Revisiting the polytopal rearrangements in penta-coordinate d7-metallocomplexes: modified Berry pseudorotation, octahedral switch, and butterfly isomerization†

Abstract
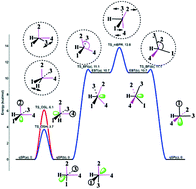
This paper provides a first-principles theoretical investigation of the polytopal rearrangements and fluxional behavior of five-coordinate d7-transition metal complexes. Our work is primarily based on a potential energy surface analysis of the iron tetracarbonyl hydride radical HFe˙(CO)4. We demonstrate the existence of distorted coordination geometries in this prototypical system and, for the first time, introduce three general rearrangement mechanisms, which account for the non-ideal coordination. The first of these mechanisms constitutes a modified version of the Berry pseudorotation via a square-based pyramidal C4v transition state that connects two chemically identical edge-bridged tetrahedral stereoisomers of C2v symmetry. It differs from the classical Berry mechanism, which involves two regular D3h equilibrium structures and a C4v transition state. The second mechanism is related to the famous “tetrahedral jump” hypothesis, postulated by Muetterties for a number of d6 HML4 and H2ML4 complexes. Here, our study suggests two fluxional rearrangement pathways via distinct types of C2v transition states. Both pathways of this mechanism can be described as a single-ligand migration to a vacant position of an “octahedron”, thus interchanging (switching) the apical and basal ligands of the initial quasi-square pyramidal isomer, which is considered as an idealized octahedron with a vacancy. Accordingly, we call this mechanism “octahedral switch”. The third mechanism follows a butterfly-type isomerization featuring a key-angle deformation, and we thus call it “butterfly isomerization”. It connects the quasi-square pyramidal and edge-bridged tetrahedral isomers of HFe˙(CO)4 through a distorted edge-bridged tetrahedral transition state of Cs symmetry. Our paper discusses the overall features of the isomers and rearrangement mechanisms as well as their implications. We rationalize the existence of each stationary point through an electronic structure analysis and argue their relevance for isolobal analogues of HFe˙(CO)4.
 Please wait while we load your content...
Please wait while we load your content...

