
Mechanism of photocatalytic water oxidation on small TiO2 nanoparticles†
 *a
Shane M.
Parker,
a
Enrico
Berardo,
bc
Martijn A.
Zwijnenburg
c
and
Filipp
Furche
*a
*a
Shane M.
Parker,
a
Enrico
Berardo,
bc
Martijn A.
Zwijnenburg
c
and
Filipp
Furche
*a
Abstract
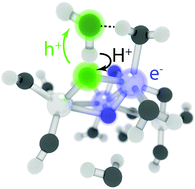
We present the first unconstrained nonadiabatic molecular dynamics (NAMD) simulations of photocatalytic water oxidation by small hydrated TiO2 nanoparticles using Tully surface hopping and time-dependent density functional theory. The results indicate that ultrafast electron–proton transfer from physisorbed water to the photohole initiates the photo-oxidation on the S1 potential energy surface. The new mechanism readily explains the observation of mobile hydroxyl radicals in recent experiments. Two key driving forces for the photo-oxidation reaction are identified: localization of the electron–hole pair and stabilization of the photohole by hydrogen bonding interaction. Our findings illustrate the scope of recent advances in NAMD methods and emphasize the importance of explicit simulation of electronic excitations.
 Please wait while we load your content...
Please wait while we load your content...

