
Inner-sphere vs. outer-sphere reduction of uranyl supported by a redox-active, donor-expanded dipyrrin†
 *a
and
Jason B.
Love
*a
*a
and
Jason B.
Love
*a
Abstract
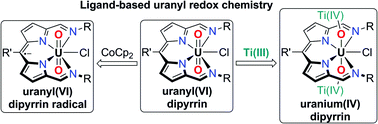
The uranyl(VI) complex UO2Cl(L) of the redox-active, acyclic diimino-dipyrrin anion, L− is reported and its reaction with inner- and outer-sphere reductants studied. Voltammetric, EPR-spectroscopic and X-ray crystallographic studies show that chemical reduction by the outer-sphere reagent CoCp2 initially reduces the ligand to a dipyrrin radical, and imply that a second equivalent of CoCp2 reduces the U(VI) centre to form U(V). Cyclic voltammetry indicates that further outer-sphere reduction to form the putative U(IV) trianion only occurs at strongly cathodic potentials. The initial reduction of the dipyrrin ligand is supported by emission spectra, X-ray crystallography, and DFT; the latter also shows that these outer-sphere reactions are exergonic and proceed through sequential, one-electron steps. Reduction by the inner-sphere reductant [TiCp2Cl]2 is also likely to result in ligand reduction in the first instance but, in contrast to the outer-sphere case, reduction of the uranium centre becomes much more favoured, allowing the formation of a crystallographically characterised, doubly-titanated U(IV) complex. In the case of inner-sphere reduction only, ligand-to-metal electron-transfer is thermodynamically driven by coordination of Lewis-acidic Ti(IV) to the uranyl oxo, and is energetically preferable over the disproportionation of U(V). Overall, the involvement of the redox-active dipyrrin ligand in the reduction chemistry of UO2Cl(L) is inherent to both inner- and outer-sphere reduction mechanisms, providing a new route to accessing a variety of U(VI), U(V), and U(IV) complexes.
- This article is part of the themed collections: Celebrating our 2018 prize and award winners and ISACS: Challenges in Inorganic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

