
Ti3BN monolayer: the MXene-like material predicted by first-principles calculations†
 *a
Lei
Liu
c
and
*a
Lei
Liu
c
and
Abstract
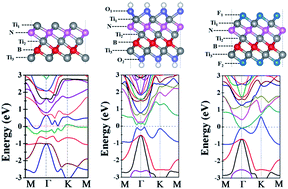
The discovery of graphene and other two-dimensional (2D) materials has set the foundation for exploring and designing novel single layered sheets. The family of 2D materials encompasses a wide selection of compositions including almost all the elements of the periodic table and they have the potential to play a fundamental role in the future of electronics, composite materials and energy technology. Therefore, searching for new 2D materials is a big challenge in materials science. In this work, we theoretically designed a monolayer of Ti3BN following the strategy of “atomic transmutation”. The Ti3BN monolayer can be considered as three Ti-atomic layers being interleaved with one N-atomic layer and one B-atomic layer, in the sequence of Ti1–N–Ti2–B–Ti3. The moderate cohesive energy, positive phonon frequencies and high melting point are the best guarantees for good stability of Ti3BN. Based on a global minimum structures search using the particle-swarm optimization (PSO) method, Ti3BN is the lowest energy structure in 2D space, which holds great promise for the realization of layered Ti3BN in experiment. Based on density functional theory (DFT) calculations, Ti3BN is intrinsically metallic and its electronic properties can be modulated by varying the surface groups, such as OH or F-termination. If realized in experiment, it may find applications in many aspects.
 Please wait while we load your content...
Please wait while we load your content...

