
Introducing DDEC6 atomic population analysis: part 3. Comprehensive method to compute bond orders†

Abstract
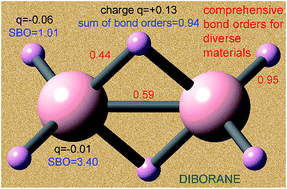
Developing a comprehensive method to compute bond orders is a problem that has eluded chemists since Lewis's pioneering work on chemical bonding a century ago. Here, a computationally efficient method solving this problem is introduced and demonstrated for diverse materials including elements from each chemical group and period. The method is applied to non-magnetic, collinear magnetic, and non-collinear magnetic materials with localized or delocalized bonding electrons. Examples studied include the stretched O2 molecule, 26 diatomic molecules, 3d and 5d transition metal solids, periodic materials with 1 to 8748 atoms per unit cell, a biomolecule, a hypercoordinate molecule, an electron deficient molecule, hydrogen bound systems, transition states, Lewis acid–base complexes, aromatic compounds, magnetic systems, ionic materials, dispersion bound systems, nanostructures, and other materials. From near-zero to high-order bonds were studied. Both the bond orders and the sum of bond orders for each atom are accurate across various bonding types: metallic, covalent, polar-covalent, ionic, aromatic, dative, hypercoordinate, electron deficient multi-centered, agostic, and hydrogen bonding. The method yields similar results for correlated wavefunction and density functional theory inputs and for different SZ values of a spin multiplet. The method requires only the electron and spin magnetization density distributions as input and has a computational cost scaling linearly with increasing number of atoms in the unit cell. No prior approach is as general. The method does not apply to electrides, highly time-dependent states, some extremely high-energy excited states, and nuclear reactions.
- This article is part of the themed collection: Computational chemistry
 Please wait while we load your content...
Please wait while we load your content...

